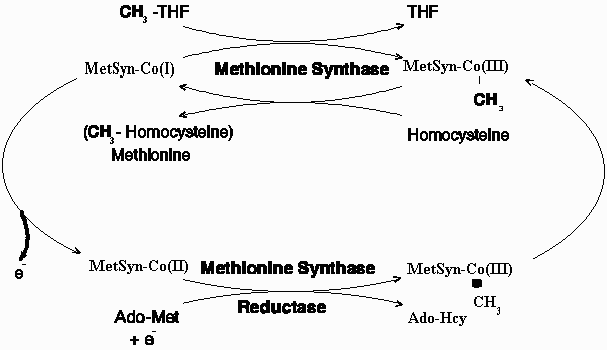
Methionine synthase catalyzes the remethylation of homocysteine to methionine via a reaction in which methylcobalamin serves as an intermediate methyl carrier. Over time, the cob(I)alamin cofactor of methionine synthase becomes oxidized to cob(II)alamin rendering the enzyme inactive. Regeneration of functional enzyme requires reductive methylation via a reaction in which S-adenosylmethionine is utilized as a methyl donor. Patients of the cblE complementation group of disorders of folate/cobalamin metabolism who are defective in reductive activation of methionine synthase exhibit megaloblastic anemia, developmental delay, hyperhomocysteinemia, and hypomethioninemia. Using consensus sequences to predicted binding sites for FMN, FAD, and NADPH, we have cloned a cDNA corresponding to the "methionine synthase reductase" reducing system required for maintenance of the methionine synthase in a functional state. The gene MTRR has been localized to chromosome 5p15.2-15.3. A predominant mRNA of 3.6 kb is detected by Northern blot analysis. The deduced protein is a novel member of the FNR family of electron transferases, containing 698 amino acids with a predicted molecular mass of 77,700. It shares 38% identity with human cytochrome P450 reductase and 43% with the C. elegans putative methionine synthase reductase. The authenticity of the cDNA sequence was confirmed by identification of mutations in cblE patients, including a 4-bp frameshift in two affected siblings and a 3-bp deletion in a third patient. The cloning of the cDNA will permit the diagnostic characterization of cblE patients and investigation of the potential role of polymorphisms of this enzyme as a risk factor in hyperhomocysteinemia-linked vascular disease.
Methionine is an essential amino acid in mammals. It is required for protein synthesis and is a central player in one carbon metabolism. In its activated form, S-adenosylmethionine, it is the methyl donor in hundreds of biological transmethylation reactions and the donor of propylamine in polyamine synthesis. The eventual product of the demethylation of methionine is homocysteine and its remethylation is catalyzed by a cobalamin-dependent enzyme, methionine synthase. The enzyme-bound cobalamin cofactor of methionine synthase plays an essential role in the methyl transfer reaction by acting as an intermediate methyl carrier between methyltetrahydrofolate and homocysteine. Cleavage of the methyl-cobalt bond of the methylcob(III)alamin intermediate occurs heterolytically so as to leave the cobalamin in the highly reactive cob(I)alamin oxidation state. During the cycling between methylcob(III)alamin and cob(I)alamin, the cofactor may be oxidized to cob(II)alamin with consequent inactivation of the enzyme. The restoration of enzyme activity is dependent on the reductive methylation of cob(II)alamin in a reaction in which S-adenosylmethionine provides the methyl group.
The E. coli methylcobalamin-dependent methionine synthase has been
well characterized. The reductive activation system required for
its maintenance is a two component flavoprotein system consisting
of flavodoxin and NADPH-ferredoxin (flavodoxin) oxidoreductase, a
member of a family of electron transferases termed the "FNR
family". Banerjee and colleagues recently documented an
NADPH-dependent reducing activity defective in cblE patients,
suggesting that the reductive-activation system in higher organisms
would bear some resemblance to that in E. coli.
Given the absence of flavodoxins in mammalian cells and the
occurrence of a single complementation group accounting for defects
of the reductive-activation system, we hypothesized that the human
counterpart would be a single protein structurally related to the
combination of flavodoxin and flavodoxin reductase.
Significantly, this would imply that FMN, FAD, and NADPH would be
bound by a single polypeptide, as is the case for cytochrome P-450
reductase and NO synthase. In this study, we present the isolation
of cDNA clones corresponding to the reductive activation enzyme
defective in the cblE complementation group. The deduced protein is
a novel member of the FNR family. We have called this enzyme
methionine synthase reductase.
click
here to get more information about cblE disease
and
here to know more about methionine synthase defiency.

click
here for a link to Daniel Leclerc Home Page.

